Chiheb Ben Mahmoud – Oxford University (UK)
Graph-based models for solid-state NMR spectroscopy
Machine learning (ML) has become an effective tool for predicting spectroscopic observables with quantum-mechanical (QM) accuracy. In this talk, I discuss perspectives on ML approaches for modeling tensorial nuclear magnetic resonance (NMR) parameters and their connection to experimentally measurable spectra.
First, I present a systematic study of graph neural network approaches for learning anisotropic magnetic shielding and electric field gradient tensors, and assess how numerical accuracy translates into experimentally relevant observables, including multidimensional spectra[1]. These methods are applied to structurally complex systems, including amorphous SiO2, zeolites, and dynamic phase transitions in crystalline materials.
Next, I introduce a data-efficient training framework based on synthetic pre-training and targeted fine-tuning. Synthetic supervision biases models toward geometry–tensor correlations, yielding up to an order-of-magnitude improvement in data efficiency in low-data regimes while preserving accuracy in the data-rich limit. I further assess chemical transferability, demonstrating meaningful transfer even to previously unseen species.
Finally, I highlight applications to zeolites[2], where ML models rained on a chemically diverse dataset enable accurate prediction of full NMR tensors and spectra for ²⁷Al, ²⁹Si, ¹⁷O, ²³Na, and ¹H, allowing direct comparison with experimental data.
These approaches illustrate how ML can accelerate the prediction and interpretation of tensorial NMR observables, bridging first-principles simulations and experimental spectroscopy for complex materials.
[1] Chiheb Ben Mahmoud, Louise A. M. Rosset, Jonathan R. Yates, Volker L. Deringer, J. Chem. Phys. 163, 024118 (2025).
[2] Carlos Bornes, Chiheb Ben Mahmoud, Volker L. Deringer, Christopher J. Heard, Lukáš Grajciar, arXiv preprint arXiv:2603.22268 [physics.chem-ph] (2026).
Giovanni Caldarelli – EPFL (CH)
Facilitating the simulation of spectroscopic features via a variational
formulation of electronic response in the presence of exchange interactions
Giovanni Caldarelli1*, Alberto Guandalini2, Francesco Macheda2, 3, and Francesco Mauri2
1Theory and Simulation of Materials (THEOS) and National Centre for Computational Design and Discovery of Novel Materials (MARVEL), École Polytechnique Fédérale de Lausanne, Lausanne
2Sapienza Università di Roma, Italy
3Università di Modena e Reggio Emilia, Italy
Accurate quantum-mechanical simulation of spectroscopic observables is essential for interpreting experimental spectra, particularly in materials governed by strong many-body and excitonic effects. State-of-the-art predictions often rely on dynamical electronic response functions derived from timedependent mean-field Hamiltonians, encompassing (screened) Hartree-Fock, Hybrid-DFT, and Bethe- Salpeter Equation (BSE) approaches. To advance the efficiency and reliability of these simulations, we present a novel variational formulation for electronic linear response functions. Traditionally, calculating these response functions utilizes a combination of screened and bare electronic vertices. We demonstrate an exact rewriting of the formalism entirely in terms of purely screened electronic vertices (‘screen-screen’)[1]. Within this framework, the response function can be evaluated as a stationary point of a functional of the exact density matrix. Consequently, any approximation of the response function yields an error that is strictly quadratic with respect to the error in the density matrix. This variational property drastically improves the efficiency and convergence of spectroscopic simulations. Crucially, we also show that the imaginary part of any electronic response— which directly dictates spectral absorption—can be rigorously cast as a generalized Fermi Golden Rule by introducing a complementary rewriting with complex-conjugated vertices (‘screen-screen’).
We showcase the practical effectiveness of our formalism by studyng a tight-biding model of graphene [2]. Despite its simplicity, we capture the strong excitonic features in the optical conductivity spectrum of graphene . Furthermore, this framework has proven successful in explaining precisely the Kohn anomaly of graphene in the presence of exchange interactions[3]. In fact, since this ‘screen-screen’ formulation is an exact mathematical rewriting of linear response theory, virtually every simulation of linear response properties stands to benefit from the formalism developed in this work, without imposing any additional computational cost in numerical implementations.
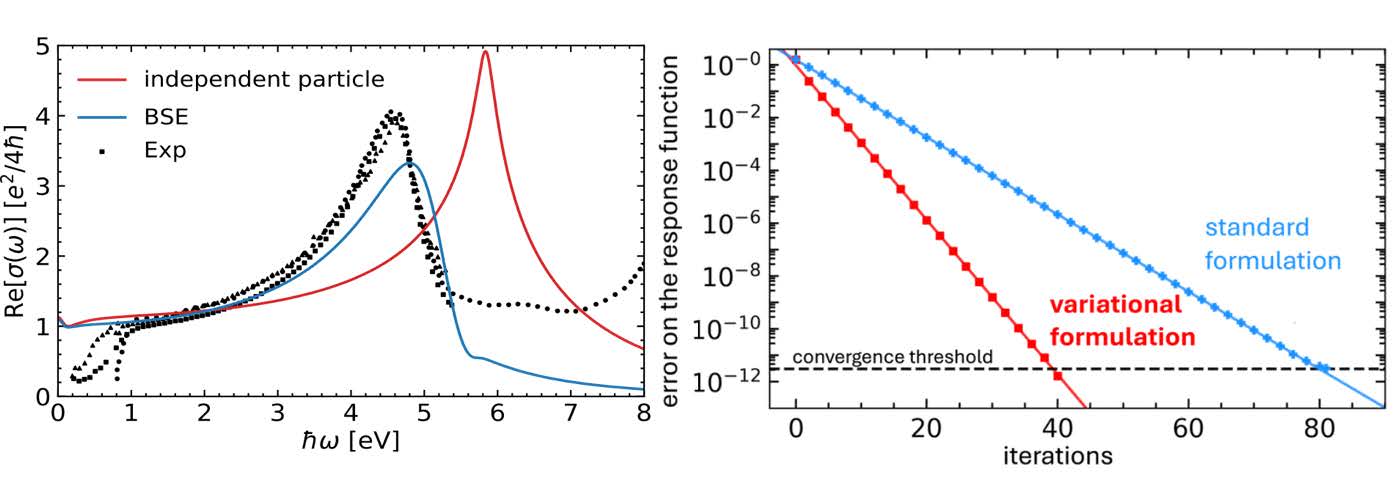
Figure 1: Our tight-binding model of graphene, we are able to reproduce the redshift of the optical conductivity of graphene due to excitonic effects (labeled BSE), and our variational formulation allow for faster and more precise iterative algorithms for the calculation of response functions.
[1] G. Caldarelli, A. Guandalini, F. Macheda, and F. Mauri, Phys. Rev. B 111, 075137 (2025).
[2] A. Guandalini, G. Caldarelli, F. Macheda, and F. Mauri, Phys. Rev. B 111, 075118 (2025).
[3] A. Guandalini, F. Macheda, G. Caldarelli, and F. Mauri, Phys. Rev. Lett. 2025, 135, 076401
David A. Egger – Technical University of Munich (DE)
Machine learning for predicting the properties of energy materials
Machine learning (ML) is reshaping computational materials science by enabling accurate predictions of properties that are difficult to access directly from first principles. In this talk, I will discuss recent developments from our group that combine physical and ML models to predict the properties of energy materials under realistic, finite-temperature conditions. First, I will introduce HAMSTER [1], a physics-informed Hamiltonian-learning framework that predicts optoelectronic properties of complex semiconductors, including halide perovskites, across temperature, composition, and system sizes. Second, I will show how ML-accelerated Raman calculations [2] reveal spectral signatures of liquid-like ionic motion in solid electrolytes, providing a route to rapidly identify fast ion conductors for solid-state batteries [3]. Finally, I will discuss equivariant neural-network models that couple ML force fields with Hamiltonian learning to describe electronic properties of semiconductor defects [4]. Together, these examples broadly demonstrate how interpretable and data-efficient ML can accelerate the discovery and understanding of functional energy materials.
[1] Schwade, Zhang, Vonhoff, Delgado, Egger, Nature Communications 17, 2652 (2026)
[2] Egger, Grumet, Bucko, J. Chem. Phys. 160, 120901 (2025)
[3] Grumet, Miyagawa, Pittet, Pegolo, Thalmann, Kaiser, Egger, AI for Science 2, 011001 (2026).
[4] Zhu, Rinke, Egger, npj Comp. Mater. 12, 169 (2026)
Mario El Kazzi – Paul Scherrer Institute (CH)
Bridging Laboratory and Synchrotron Operando Spectroscopies for Multiscale Battery Interface insights
Mario El Kazzi,1,* Valerie Siller1, Barthélémy Lelotte1, and Carlos A. F. Vaz2
1PSI Center for Energy and Environmental Sciences, Paul Scherrer Institute, PSI, Switzerland.
2PSI Center for Photon Science, Paul Scherrer Institute, PSI, Switzerland.
All-solid-state batteries (ASSBs) promise high energy density and improved safety, yet their performance is limited by complex interfacial reactions and chemo-mechanical degradation at buried solid-solid interfaces. Gaining a fundamental understanding of these dynamic processes requires operando pectroscopic techniques capable of probing chemical and electronic changes in real time [1]. In this talk, we highlight recent advances in operando spectroscopies, combining laboratory-based X-ray hotoelectron spectroscopy (XPS) [2, 3] with synchrotron techniques such as X-ray absorption spectroscopy (XAS)[4] and X-ray photoemission electron microscopy (XPEEM) [5, 6]. Dedicated electrochemical cells enable reliable measurements under realistic conditions while maintaining compatibility with photon-based probes. The complementarity between in-house and synchrotron approaches provides access to surface and bulk sensitivity, as well as spatially resolved chemical information.
Using sulfide-based ASSBs as model systems, operando XPS reveals electrolyte decomposition pathways and the formation of interphase species, while operando XAS tracks transition-metal redox and oxygen activity in layered oxide cathodes. Nanoscale XPEEM further resolves local heterogeneities and the role of surface coatings in stabilizing interfaces.

Fig. 1: Schematic overview of the in-house and synchrotron-based X-ray spectroscopy approaches employed to probe all-solid-state batteries across multiple length scales. Different cell configurations adapted to each technique, emphasizing their compatibility with ultra-high vacuum and photon-based probes.
[1] K.V. Kravchyk et al. Chimia 78 (6), 403-414 (2024).
[2] V. Siller et al., Small 21 (46), e08796 (2025).
[3] X. Wu et al. ACS Appl. Mater. Interfaces 13 (36), 42670-42681 (2021).
[4] B. Lelott, et al., ACS Applied Materials & Interfaces 17 (9), 14645-14659 (2025).
[5] B. Lelotte et al., Surfaces and Interfaces, 73, 107490 (2025).
[6] M. Mirolo et al., Analytical Chemistry 92 (4), 3023-3031 (2020).
Claudia Fasolato – University of Rome “La Sapienza” (IT)
Time-resolved Raman spectroscopy in photoexcited MoS2: transient doping, resonance effects and electron-phonon coupling
MoS2 belongs to the class of graphene-like, layered materials called transition metal dichalcogenides (TMDs). Semiconducting TMDs, like MoS2, show an indirect to direct bandgap transition when the system is exfoliated down to the monolayer structure, which makes them promising materials for future applications in optoelectronics and related fields [1]. Unravelling the relaxation pathways of photoexcited electrons in such systems, and specifically their coupling with the lattice degrees of freedom, is of great applicative interest.
Time-resolved Raman spectroscopy (TRRS) can provide direct access to the electron-phonon coupling dynamics, as well as the incoherent phonon relaxation [2]. The aim of our experiment is studying the deexcitation dynamics of MoS2 after impulsive optical pumping of the excitonic bandgap. At the SPRINT-NFFA facility, we have carried out a TRRS study on the system, monitoring the temporal evolution of two specific Raman active features, namely the out-of- plane A1g mode and the in-plane E’2g mode, after the photoexcitation. Equilibrium Raman spectroscopy studies have linked the modification of such phonon lineshapes to doping effects, providing the background for studying the optically induced, transient doping of the system [3,4,5]. To explore the effect of dimensionality on the system relaxation dynamics, the experiment was performed on both bulk and monolayer MoS2, revealing significant differences in the phenomenology and relaxation timescales. The role of the substrate on the monolayer deexcitation dynamics was explored by selecting substrates with different electrical and thermal properties. After presenting the results of the TRRS study [6], I will discuss their perspective integration in a multi-messenger, time-resolved spectroscopy study, coupling the time resolved photon and electron spectroscopies available at the SPRINT-NFFA facility (CNR IOM @Elettra, Trieste, Italy).
[1] X. Zhang et al., Chem Soc Rev, Vol. 44, No. 9, 2757 (2015).
[2] S. Han et al., Optics Express, Vol. 27, No. 21, 29949 (2019).
[3] G. Kukucska et al., Physica Status Solidi B, Vol. 254, No. 11, 1700184 (2017).
[4] B. Chakraborty et al., Phys Rev B, Vol. 85, No. 16, 161403 (2012).
[5] M. Yamamoto et al., J Phys Chem C, Vol. 117, No. 48, 25643 (2013).
[6] A. M. Finardi et al., npj 2D Materials and Applications, Vol. 9, No. 1, 79 (2025).
Myrta Grüning – Queen’s University Belfast (UK)
Nonlinear optics from first-principles real-time approaches
Myrta Grüning1*, Claudio Attaccalite2
1Queen’s University Belfast– Belfast, Northern Ireland
2CINAM-CNRS – Marseille, France
Nonlinear optical techniques are offering access to ultrafast dynamics, symmetry properties, and light- matter interactions that go beyond the linear regime. However, accurately predicting nonlinear optical signals from theory remains challenging: these responses often depend on subtle electron-electron and electron-hole interactions that are not captured by independent particle calculations.
The GW approximation and the Bethe Salpeter equation-computational many-body techniques have become central tools for describing excited state and optical properties of solids [1]. These computational techniques were originally developed for linear spectroscopy. In Refs.[2,3], we put forward a first- principles approach that extends these approaches to nonlinear optical regimes and thus provides predictive insight into nonlinear optical phenomena in dielectric materials. After introducing the key ideas behind the approach, I will present applications to second- and third-harmonic generation, two-photon absorption [4], shift-current [5] and frequency-mixing spectroscopies [6].
[1] G. Onida, L. Reining, and A. Rubio. Reviews of Modern Physics, 74, 601-659 (2022).
[2] M. Grüning and C. Attaccalite. Physical Review B 89, 081102 (2014).
[3] C. Attaccalite and M. Grüning. Physical Review B 88, 235113 (2013).
[4] C. Attaccalite, M. Grüning et al., Phys. Rev. B 98, 165126 (2018).
[5] Y. Mao, J. Zhou, M. Grüning, and C. Attaccalite. Phys. Rev. Materials 9, 124002 (2025).
[6] M. N. Pionteck, M. Grüning, S. Sanna, and C. Attaccalite. SciPost Phys. 19, 129 (2025).
Alexei Kuzmin – University of Latvia (LV)
Machine learning for X-ray absorption spectroscopy
Alexei Kuzmin*, Inga Pudza, Pjotrs Žguns
Knowledge of material structure is required to predict, understand, and optimize material properties. X-ray absorption spectroscopy (XAS) is a local structural method [1] that is widely used nowadays due to the accessibility of synchrotron radiation sources and the availability of commercial laboratory setups. XAS can probe the local environment of a desired chemical element in both bulk and diluted materials with ordered or disordered structures under various experimental conditions. The theoretical foundations of X-ray absorption spectra are well established [2, 3]; however, their interpretation remains a complex task, especially in multicomponent compounds [4], due to the ill-posed nature of the problem. State-of-the-art approaches to XAS data analysis (Figure 1) are based on atomistic simulations [5], such as molecular dynamics (MD) or reverse Monte Carlo methods. Besides, over the last ten years, efficient machine learning (ML) methods have started to emerge for solving forward and inverse problems, i.e., predicting spectroscopic data from material structure and extracting structural information from experimental spectra.
In this presentation, examples of ML applications in XAS will be given. In particular, the use of ML interatomic potentials for predicting X-ray absorption spectra based on the results of MD simulations will be discussed [6]. The application of an artificial neural network for reconstructing atomic structures during a phase transition will also be presented.

Figure 1: A workflow for structure determination using X-ray absorption spectroscopy.
[1] A. Kuzmin and J. Chaboy. IUCrJ 1, 571 (2014).
[2] J. J. Rehr, and R. C. Albers. Rev. Mod. Phys. 72, 621 (2000).
[3] C. R. Natoli, P. Krüger, K. Hatada, K. Hayakawa, D. Sébilleau, and O. Šipr. J. Phys.: Condens. Matter 24, 365501 (2012).
[4] A. Kuzmin, G. Yasin, M. Abubaker Khan, M. Afifi, T. Anh Nguyen, and Y. Zhang. X-ray absorption spectroscopy in high-entropy material research. High-Entropy Alloys: Design, Manufacturing, and Emerging Applications, eds. Elsevier (2024).
[5] J. Timoshenko, A. Kuzmin, C. Chantler, F. Boscherini, and B. Bunker. Reverse Monte Carlo and molecular-dynamics approaches to EXAFS analysis. International Tables for Crystallography, Volume I: X-ray Absorption Spectroscopy and Related Techniques (2024).
[6] P. Žguns, I. Pudza, and A. Kuzmin. J. Chem. Theory Comput. 21, 8142 (2025).
Frederico Alves Lima – European XFEL (DE)
Ultrafast time-resolved X-ray spectroscopies at X-ray Free-electron Lasers
F. Lima*, Y. Uemura, X. Huang, M. Biednov, H. Yousef, S. Hayes, A. Sarkar, D. Bregenholt Jakobsen, H. Wang, H. Xu, M. Knoll, P. Frankenberger, S. Huynh, D. Khakhulin, P. Zalden, and C. Milne.
X-ray spectroscopies are widely used in the investigation of materials, providing detailed information about the electronic configuration and local structures around the absorbing element [1, 3]. In the hard X-ray regime (> 4.5 keV) they probe transition metals, with countless applications in materials’ science, physics, chemistry, biology, geology, etc [1, 3]. Common to all variations of X-ray spectroscopy is the absorption of a high energy X-ray photon by a core electron, which is then ejected from the absorbing element and propagates in the material in the form of a photoelectron. X-ray Absorption Spectroscopy (XAS) explores this phenomenon, producing spectra that delivers distinct information depending on the energy of this photoelectron. Following the X-ray absorption process, the absorbing element is left with a hole in one of its core levels, which is quickly refilled and an X-ray photon is emitted. This forms the basis of X-ray emission Spectroscopy (XES), which also has different sensitivity depending which electronic levels are involved in the refilling process [2, 3]. Combining XAS and XES in a single experiment, called resonant XES (RXES) leads to improved information content [2, 3].
Over the last decades, X-ray spectroscopies have advanced significantly with the increased availability of X-ray Free-electron Laser (XFEL) sources [2]. Thanks to the unprecedented brilliance and short X-ray pulses provided by XFELs, the implementation of X-ray spectroscopies with <100 fs resolution is now a reality, leading to major breakthroughs in ultrafast science [2]. The FXE instrument at the European XFEL stands out providing state-of-the-art capability for performing a variety of X-ray spectroscopy experiments with ultrafast time resolution [4-7]. The large suite of high-resolution X-ray spectrometers available at FXE, combined with the high repetition rate operation of European XFEL allows a variety of time-resolved X-ray spectroscopy experiments to be performed. Examples include a recent implementation of an X-ray spectrometer optimized for high photon energies [7] and measurements of extended range XAS (also known as EXAFS) with ~100 fs resolution. In this contribution I will introduce the instrumentation currently installed at FXE and present a few cases of ultrafast X-ray spectroscopies. Some data will be discussed in the context of computer-aided spectroscopy.
[1] Koningsberger, D. C. & Prins, R. X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES. (John Wiley & Sons, 1988).
[2] Bergmann, U. et al. Nature Reviews Physics 3, 264–282 (2021).
[3] Cutsail, G. E., III & DeBeer, S., ACS Catal. 12, 5864–5886 (2022).
[4] Galler, A. et al., Journal of Synchrotron Radiation 26, 1432–1447 (2019).
[5] Khakhulin, D. et al., Applied Sciences 10, 995 (2020).
[6] Lima, F. A. et al. Journal of Synchrotron Radiation 30, 1168–1182 (2023).
[7] Huang, X. et al. Journal of Synchrotron Radiation 32, 506–523 (2025).
Johannes Lischner – Imperial College London (UK)
Probing metal oxide surface chemistry with core-electron binding energies
X-ray photoemission spectroscopy (XPS) is a powerful technique to gain insight into the chemical properties of oxide surfaces. However, the interpretation of XPS spectra is notoriously difficult as realistic surfaces contain different terminations, reconstructions, adsorbates and defects all of which leave (potentially overlapping) spectroscopic fingerprints. To address this challenge, I will present a first-principles approach based on the Z+1 method that allows us to predict XPS spectra of oxide surfaces which can directly be compared to experimental measurements. We present results for different SnO2 (110) surfaces: the stoichiometric surface, surfaces with different types of vacancies (one of which is the fully reduced surface) and also the fully reduced surface with adsorbed OH and O2 molecules. For these systems, we calculate the O 1s core-electron binding energies of all oxygen atoms and then use this to predict the XPS spectrum. We find that the fully reduced surface gives rise to a highly symmetric peak shape in agreement with recent XPS measurements. In contrast, the spectrum of the stoichiometric surface exhibits an additional feature at low binding energies caused by the bridging oxygen atoms at the surface. For the reduced surface with OH and O2 adsorbates, the spectrum exhibits additional features at higher binding energies. The predicted spectra are in good agreement with experimental results obtained for reduced surfaces that have been exposed to oxygen gas. The presented method is general and can be straightforwardly applied to other surfaces.
Laura Ratcliff – University of Bristol (UK) and UiT The Arctic University of Norway,
Tromsø (NO)
Exploring Disorder using Density Functional Theory and X-ray Photoelectron Spectroscopy
While theoretical simulations are routinely performed for interpreting valence X-ray photoelectron spectroscopy (XPS), the interpretation of core XPS primarily relies on comparison to reference spectra. This is challenging, if not impossible, in the case of large molecules or complex solids containing many different local chemical environments, even more so for systems exhibiting disorder. Recently, however, theoretical approaches for calculating core binding energies (BEs) are becoming increasingly common. One approach, based on density functional theory, which has shown great promise for core BE calculations, is $\Delta$SCF. However, its use in Gaussian basis codes is complicated by the need to use either very large or specifically constructed basis sets which are adapted for core-excited calculations. At the same time, such codes may also suffer from problems such as core-hole hopping, which can lead to poor convergence or inaccurate results. In this talk I will describe how an adaptive multi-wavelet basis can be employied to overcome these problems [1,2]. When combined with a plane-wave basis set approach using core-hole pseudopotentials, it becomes possible to calculate core BEs of both molecules and solids using systematic basis sets, enabling core BE calculations of systems ranging from amino acids [1] to disordered molecular [3] and solid state materials [4], offering new opportunities for interpreting core XPS of complex and disordered materials.
1] J. M. Pi, M. Stella, N. K. Fernando, A. Y. Lam, A. Regoutz, and L. E. Ratcliff. J. Phys. Chem. Lett. 11, 2256 (2020).
[2] N. Göllmann, M. R. Ludwig, P. Wind, L. E. Ratcliff and L. Frediani. Phys. Chem. Chem. Phys. 27, 23013 (2025).
[3] N. K. Fernando, M. Stella, W. Dawson, T. Nakajima, L. Genovese, A. Regoutz, and L. E. Ratcliff. Phys. Chem. Chem. Phys. 24, 23329 (2022).
[4] L. E. Ratcliff, T. Oshima, F. Nippert, B. M. Janzen, E. Kluth, R. Goldhahn, M. Feneberg, P. Mazzolini, O. Bierwagon, C. Wouters, M. Nofal, M. Albrecht, J. E. N. Swallow, L. A. H. Jones, P. K. Thakar, T.-L. Lee, C. Kalha, C. Schlueter, T. D. Veal, J. B. Varley, M. R. Wagner, and A. Regoutz. Adv. Mater. 34, 2204217 (2022).
Michiel J. van Setten – IMEC (BE)
Unraveling the photo resist chemistry of EUV lithography
Photolithography is the cornerstone technology enabling the rapid, reliable, and cost-effective production of microchips. Through a sophisticated optical system, light patterns are generated to define the structures required for transistor fabrication on silicon wafers. These patterns are transferred into a physical form by inducing a solubility switch in a photoresist layer. This patterned layer serves as a mask in the subsequent etching step to transfer the pattern into the underlying silicon. To achieve feature sizes close to ten nanometers, extreme ultraviolet (EUV) lithography is required and represents the current state of the art. Attaining ultimate pattern fidelity while minimizing line-edge roughness critically depends on precise optimization of the photoresist materials. However, optimizing them by unraveling the chemical reactions that are initiated by the absorption of the 92 eV photons in these amorphous, multi-component thin films remains a significant challenge.
At imec’s AttoLab, we investigate these processes by intertwining in situ photoemission spectroscopy and computational spectroscopy. By exposing the resist with incremental, low-dose steps and using the same photons for electron emission spectroscopy, we obtain a detailed, dose-resolved view of the evolving spectral signatures during exposure. Systematic comparison of the measured spectra with first principles simulated spectra enables us to directly link observed spectral changes to the underlying atomistic processes.
In this presentation, we will highlight the integrated methodology used to generate structural models and compute the corresponding spectra, including GW embedding approaches for core-level spectroscopy and force-based Monte Carlo structure optimization. We will illustrate the power of this framework with two case studies on distinct classes of resist materials, demonstrating how this combined experimental–computational approach leads to new and unexpected insights.
Tigany Zarrouk – Aalto University (FI)
Molecular Augmented Dynamics: Generating experimentally consistent
atomistic structures by design
A fundamental objective of materials modeling is identifying atomic structures that align with experimental observables. Conventional approaches for disordered materials involve sampling from thermodynamic ensembles and hoping for an experimental match. This process is inefficient and offers no guarantee of success. We present a method based on modified molecular dynamics, that we call molecular augmented dynamics (MAD) [1, 2], which identifies structures that simultaneously match multiple experimental observables and exhibit low energies as described by a machine learning interatomic potential (MLP) trained from ab-initio data. We demonstrate its feasibility by finding representative structures of glassy carbon, nanoporous carbon, ta-C, a-C:D and a-COx that match their respective experimental observables—X-ray diffraction, neutron diffraction, pair distribution function and X-ray photoelectron spectroscopy data (see Fig. 1)—using the same initial structure and underlying MLP. The method is general, accepting any experimental observable whose simulated counterpart can be cast as a function of differentiable atomic descriptors, as can be seen for IR and Raman spectroscopy formulations. This method greatly increases the system sizes of atomic structures that can be inferred from experimental data, in comparison to previous Monte Carlo approaches for structural inference [3].
[1] T. Zarrouk and M. A. Caro, “Molecular augmented dynamics: Generating experimentally consistent atomistic structures by design”, ArXiv preprint, arxiv.org/abs/2508.17132, (2025).
[2] T. Zarrouk and M. A. Caro, “Linear-scaling calculation of experimental observables for molecular augmented dynamics simulations”, ArXiv preprint, arxiv.org/abs/2509.22388, (2025).
[3] T. Zarrouk, R. Ibragimova, A. P. Bartók and M. A. Caro, “Experiment-Driven Atomistic Materials Modeling: A Case Study Combining X-Ray Photoelectron Spectroscopy and Machine Learning Potentials to Infer the Structure of Oxygen-Rich Amorphous Carbon”, J. Am. Chem. Soc., 146, 21, (2024).